







WELCOME TO TRUFUSION CLINICS
Ignite Your Glow
At TruFusion Clinics, located in the heart of Vacaville, CA, we are more than just a med spa; we are your trusted partner in the journey toward optimal health and wellness. Our mission is to revolutionize personalized wellness by offering a comprehensive approach that transcends traditional healthcare.
Now Introducing Our Newest Addition
The Award-Winning Multi-Application Laser System
Fotona SP Dynamis
WELCOME TO TRUFUSION CLINICS
Ignite Your Glow
At TruFusion Clinics, located in the heart of Vacaville, CA, we are more than just a med spa; we are your trusted partner in the journey toward optimal health and wellness. Our mission is to revolutionize personalized wellness by offering a comprehensive approach that transcends traditional healthcare.
Our Services
TruFusion Clinics specialists offer extensive services personalized to your unique needs and objectives.

TightSculpting®
Our TightSculpting® is a unique dual-wavelength, non-invasive laser treatment for sculpting and skin tightening on all body areas. Tighten your stomach, arms, thighs and more trouble-free areas.

Vaginal Rejuvenation
Our IntimaLase® treatment provides a non-surgical, laser-based solution for vaginal tightening, enhancing vaginal health, and improving the overall quality of life for women.

Acne & Acne Scar Revision
We offer a laser acne treatment protocol that provides a truly comprehensive solution to the problem of acne.

Fotona4D®
Achieve skin tightening and volumization without fillers or toxins, enjoy a comprehensive lifting treatment from the inside out, and see immediate rejuvenation, tightening, and glow without the need for surgery.

NightLase® Snoring and Apnea Treatment
A non-invasive, patient-friendly laser treatment for increasing the quality of a patient’s sleep. NightLase reduces the effects of sleep apnea and decreases the amplitude of snoring.

LipLase®
Non-invasive plumping for fuller lips without Injectables. There is nothing artificial as patients develop their own collagen in their lips.

SmoothEye®
SmoothEye® is a new and exciting non-ablative Fotona SMOOTH® mode treatment for tightening of the periocular region and reducing the appearance of periorbital wrinkles.

Laser Podiatry
Our Fotona Laser Podiatry treatments offer advanced, non-invasive solutions for various foot conditions, promoting faster healing and enhanced patient comfort.

Permanent Hair Reduction
Safe and effective hair reduction – using an innovative system that effectively targets hair follicles with selective and homogenous photothermolysis.

LineLase™
Our Minimally Invasive Laser Treatment is effective, safe for stretch marks, and appropriate for all skin types.
Wellness

Wellness Infusion Therapy
You can transcend your traumas and let go of obstacles on every level of physical, psychological, and spiritual blockages with our IV Wellness Infusions.

Medical Aesthetics
Experience cutting edge Medical Aesthetics to achieve your youthful glow and transformation
Medical Aesthetics & Skin Health
We offer a range of advanced medical aesthetics services designed to help you look and feel your best. Our cutting-edge treatments are tailored to meet your unique needs, ensuring optimal results and enhanced skin health.
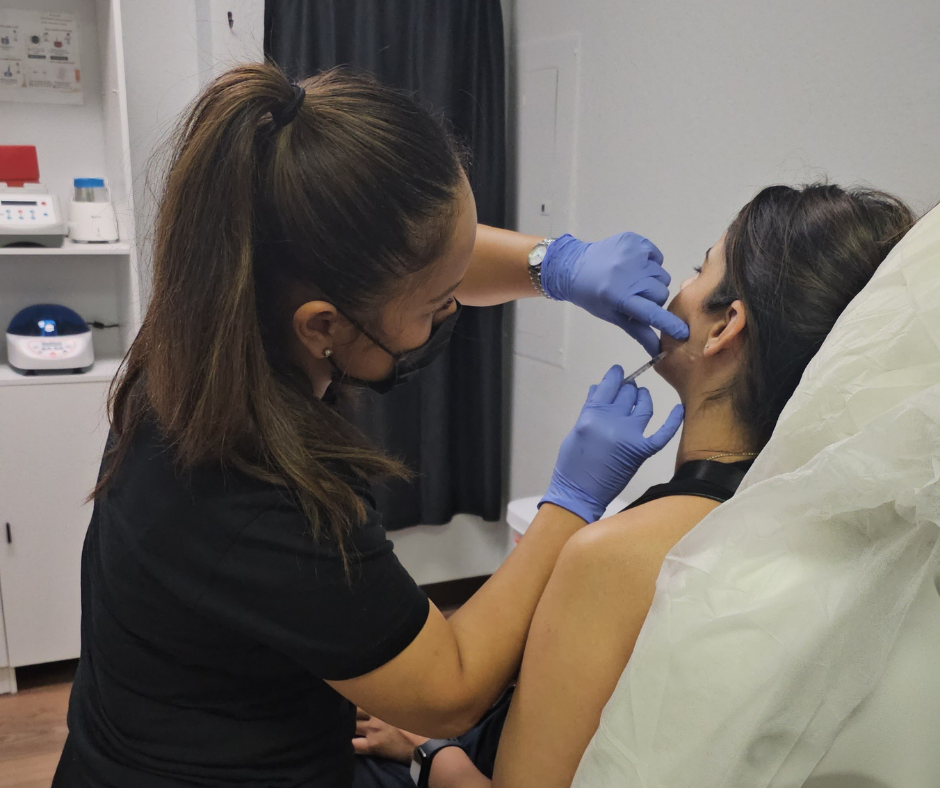
Neurotoxin Treatments
Experience the transformative effects of our Neurotoxin Treatments. By targeting fine lines and wrinkles, these treatments relax facial muscles to create a smoother, more youthful appearance. Whether you're looking to soften crow's feet, frown lines, or forehead wrinkles, our skilled practitioners will deliver natural-looking results that boost your confidence.
Reduces fine lines and wrinkles
Creates a smoother, more youthful appearance
Quick, minimally invasive procedure
Natural-looking results
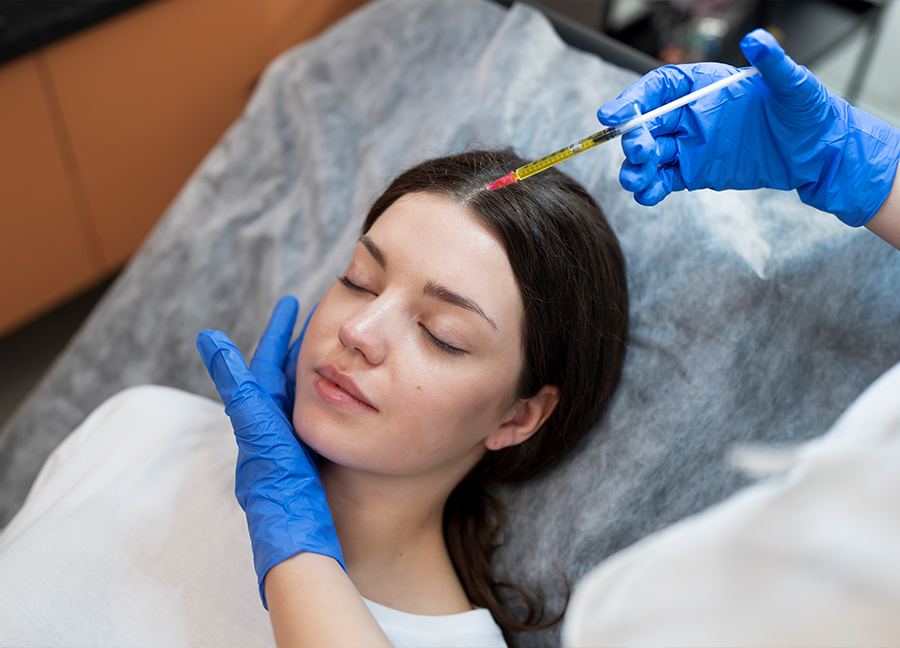
Revolutionize Your Skincare with PRF Facials
Rejuvenate your skin with our PRF (Platelet-Rich Fibrin) Facials. This innovative treatment uses your body's own growth factors to promote collagen production, improve skin texture, and reduce signs of aging. PRF Facials are a safe and effective way to achieve radiant, youthful skin without synthetic fillers or chemicals.
Uses natural growth factors
Promotes collagen production
Improves skin texture
Reduces signs of aging
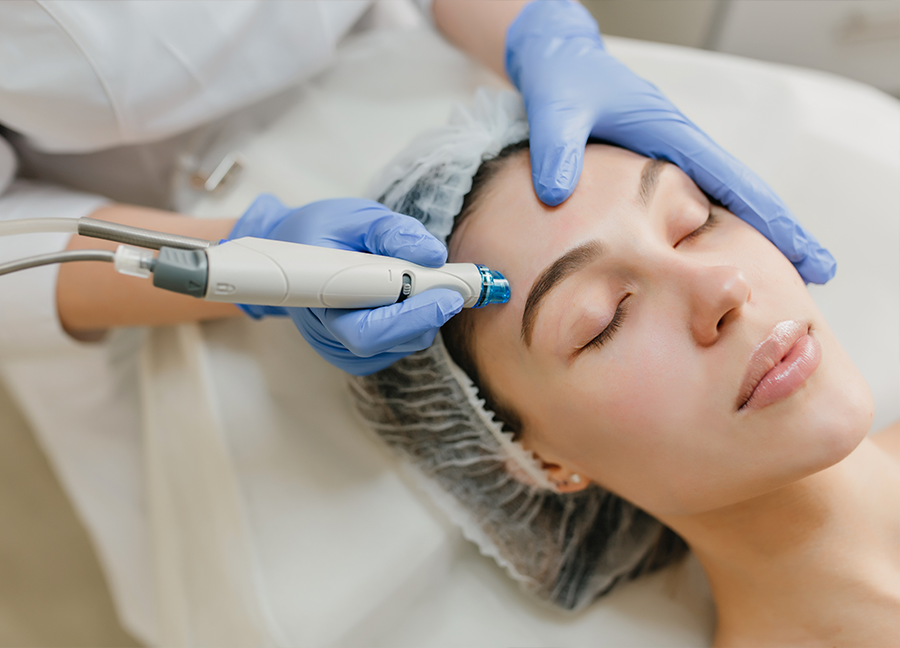
Stimulate Skin Renewal with Microneedling
Our Microneedling treatments stimulate your skin's natural healing process, promoting collagen and elastin production. This minimally invasive procedure helps to reduce the appearance of scars, fine lines, and uneven skin tone. With minimal downtime, Microneedling is an excellent option for those seeking to enhance their skin's overall texture and firmness.
Stimulates collagen and elastin production
Reduces scars, fine lines, and uneven skin tone
Minimally invasive with minimal downtime
Enhances overall skin texture and firmness

V Carbon Peel
Discover the benefits of the V Carbon Peel, a powerful skin rejuvenation treatment that combines the exfoliating properties of carbon with advanced laser technology. This treatment effectively removes impurities, reduces pore size, and evens out skin tone, leaving you with a brighter, more youthful complexion. Ideal for all skin types, the V Carbon Peel is a quick and painless way to achieve glowing skin.
Combines carbon exfoliation with laser technology
Removes impurities and reduces pore size
Evens out skin tone
Suitable for all skin types
Quick and painless procedure
Testimonials

“I visited TruFusion Clinics for facial rejuvenation, and I’m beyond satisfied with the results. My skin is glowing and feels healthier than ever. The staff is attentive and answered all my questions. Plus, the clinic has such a soothing atmosphere. Can’t wait for my next appointment!”
Sophia R
Vacaville, CA
“The team at TruFusion is exceptional. I’ve tried their IV Drip Therapy and Injectables, and the results were fantastic. The staff is professional, knowledgeable, and made me feel comfortable throughout the process. Highly recommend!”
Devon R
Fairfield, CA
“I’ve always been hesitant about IV therapy, but the team at TruFusion Clinics put me at ease. I tried their IV Glutathione treatment, and I’ve never felt more energized! The staff was so friendly and professional. I highly recommend their services.”
Mabel R
Suisun, CA
“TruFusion Clinics have changed my life with their IV Ketamine Therapy. I had been struggling with ptsd and chronic pain for years, and this treatment has given me a new lease on life. The team is professional, caring, and truly dedicated to improving your health and well-being.”
Chris F
Fairfield, CA
Book An Appointment
©Trufusionclinics 2023